
|

|

Abstract
An interactive computer procedure is described which determines 1H-1H couplings from fitting the cross peak multiplets in regular phase-sensitive COSY spectra. Robustness and simplicity of the method relies on the fact that a given cross peak intensity is not an independent variable in the fitting procedure, making it possible to measure couplings accurately even from individual cross peaks with unresolved multiplet structure.
Introduction
1
H-1H J couplings provide important dihedral information, which is widely used during NMR structure determination. Numerous methods have been proposed for measuring such couplings, primarily aimed at cases where the 1H multiplet is too overlapped or broad in the 1D spectrum to yield resolvable splittings. These include homonuclear J-spectroscopy [Aue, 1976 #2386], homonuclear E.COSY methods [Griesinger, 1987 #806], heteronuclear E.COSY [Willker, 1992 #3143], triple resonance E.COSY [Montelione, 1989 #2389], quantitative J correlation [Vuister, 1993 #215; Grzesiek, 1995 #90], comparison of cross sections through in-phase and antiphase cross peaks [Titman, 1990 #2391; Prasch, 1998 #2360], and the so-called DISCO method [Kessler, 1985 #2392], which relies on the same principle. Two new quantitative J correlation methods, based on constant-time COSY can also yield useful information [Tian, 2000 #3137; Wu, - #3152]. None of these approaches is fully satisfactory, however, when working with macromolecules at natural abundance.Here, we describe a simple but effective method to directly obtain quantitative J-coupling information from cross peaks in regular phase-sensitive COSY spectra by amplitude-constrained multiplet evaluation (ACME). Problems with deriving J splittings from antiphase COSY spectra have long been recognized, [Neuhaus, 1985 #3147; Smith, 1991 #3146; Ludvigsen, 1991 #3145] and relate to the fact that the antiphase peak-to-peak splitting in a COSY type spectrum is a function of both the line width and the magnitude of the passive and active J couplings. Some methods derive the J couplings from least squares fitting of a cross peak to a convolution of an antiphase and several in-phase splittings, using the intensity of the cross peak as a variable, non-constrained parameter in the fit. However, an increase in line width results in a larger antiphase separation of lower intensity for the cross peaks, whereas an increase in active unresolved J coupling results in a similar increase in antiphase splitting but larger cross peak intensity. Without constraining the amplitude of the cross peak, this makes it difficult to separate the effects of line width and active coupling, resulting in poorly determined J values.
Inherently better algorithms fit either a group of multiplets or the entire NMR spectrum simultaneously [Madi, 1988 #3149][Yang, 1994 #3208; Yang, 1994 #3207; Schmidt, 1998 #3206]. These algorithms implicitly use the fact that all fitted multiplets have the same intrinsic intensity (i.e., the same integrated intensity if all splittings were in-phase). The effectiveness of these methods is due to the fact that active coupling values in one multiplet appear as passive couplings in related multiplets. Typically, such methods require extensive definitions of the spin topologies and chemical shift assignments of all related multiplets, and may require some degree of multiplet fine structure. Furthermore, because of the potentially large numbers of spectral parameters to be fit, these methods usually require prior knowledge or tight constraints for many spectral parameters in order to make optimization tenable. This has so far limited the application of these methods.
Here, we demonstrate that by simply constraining the multiplet intensity, the ACME method makes it possible to extract couplings accurately and conveniently by fitting individual multiplets or small clusters of overlapping multiplets, without the need to consider all related multiplets simultaneously. To establish the multiplet intensity constraint, we use the information that the intrinsic intensity of all multiplets in the spectrum is the same, and identical to that of the in-phase diagonal multiplets for the case where a 90û mixing pulse is used. This procedure is very simple from a user perspective and circumvents the convergence problem, while retaining a sharp and accurate minimum for the fitted active J coupling. This method does not require fine structure for the antiphase multiplets that need to be fitted, and therefore is also ideally suited to fitting the very complex unresolvable multiplets that are typically obtained in COSY spectra of macromolecules weakly oriented in a liquid crystalline phase.
Ignoring cross-correlated transverse relaxation, and assuming the weak coupling limit, the time domain signal of the A-spin diagonal, SAA(t1,t2), and AX cross peak, SAX(t1,t2), in a COSY experiment are given by:
SAA(t1,t2) = So Pk cos(pJAk t1) cos(WAt1) exp(-t1/T2A)
• P
k cos(pJAkt2) exp(iWA t 2) exp(-t2/T2A) [1a]SAX(t1,t2) = So sin(pJAX t1) Pk¼ X cos(pJAk t1) cos(WAt1) exp(-t1/T2A)
•
sin(pJAXt2) Pq¼ A cos(pJXqt2) exp(iWX t 2) exp(-t2/T2X), [1b]where the products extend over all spins k coupled to A, and spins q coupled to X, WA and WX are angular chemical shifts of spins A and X, and T2A and T2X are the A- and X-spin transverse relaxation times. It is clear from Eq. [1b] that the initial cross peak "buildup" in the (t1,t2) time domain is simply So sin(pJAX t1) sin(pJAX t2) ™ So p2 JAX2t1t2. So, if So is known, the buildup of the time domain A-X cross peak signal, which simply can be obtained by inverse Fourier transformation of a resolved cross peak multiplet, provides a unique value for JAX, with effects from passive couplings and T2 only occurring at later times. As a result, JAX is an orthogonal variable relative to both the passive couplings and T2, but parallel to So. It is therefore critical that an accurate value for So is obtained prior to fitting the cross peaks. If the delay between scans is sufficiently long, the same So value applies for all multiplets within a given molecule and can be obtained from the initial (t1 = t2 = 0) time domain amplitude of either a diagonal multiplet (Eq.[1b]), or the entire normalized time domain signal. The need for constraining the amplitude (scale factor) in the fit has been mentioned before [Madi, 1988 #3149][Yang, 1994 #3208; Yang, 1994 #3207][Schmidt, 1998 #3206], but was less critical in the application to fitting of the fine structure of multiple related multiplets of a given spin system. So, the main difference relative to earlier fitting procedures is that in ACME fitting the intrinsic signal intensity, So, is held constant for all multiplets in the spectrum at a value determined experimentally from the diagonal (see below), and fine structure of the multiplet is not required for accurate measurement of a coupling. In contrast to most other fitting procedures, only the fitted value for the active coupling is meaningful. Fitted passive couplings and decay rates are parallel variables in parameter space and their optimum values generally do not provide useful coupling information. In practice, the actual fit is carried out in the frequency domain, using multiplet models generated by numerical Fourier transformation of the model time domain forms of Eq. 1. The numerical Fourier processing of the model function is performed automatically during the fit according to the zero filling and window functions that were applied to the experimental data.
The fitting procedure has been implemented via a graphical interface constructed using NMRWish [Cornilescu, 1999 #60], a companion package to the NMRPipe processing and analysis system [Delaglio, 1995 #63]. NMRWish is a version of the Tcl/Tk script interpreter "wish" [Ousterhout, 1994 #3154; Johnson, 1994 #2675], which has been augmented to include facilities for spectral display and manipulation, as well as relational database functions for manipulating spectral parameters, molecular structures, etc. Use of the graphical interface to extract couplings typically involves the following steps:
The fitting procedure itself is presently implemented via a macro interpreter that generates the model function at each iteration. This allows for substantial flexibility, for example by making it straightforward to adjust the model to account for dephasing delays inserted prior to the acquisition, or to extend the method to 3D data. However, use of an interpreted fitting function makes this implementation relatively slow, with computation times of several seconds for fitting a single multiplet, and a minute or more for complicated clusters of multiplets. Nevertheless, this is still fast enough for the method to be convenient, especially in light of the fact that no complicated definition or set-up is required in order to extract couplings. More information is provided at https://spin.niddk.nih.gov/bax-apps/NMRPipe/acme.
Prior to fitting the cross peaks, it is useful to remove the diagonal signals, which as a result of their long dispersive tails can interfere with the cross peaks. This can simply be done by subtracting the diagonal region of the COSY spectrum where the diagonal is phased to be absorptive, followed by Hilbert transformation and rephasing to make the cross peaks absorptive [Pelczer, 1991 #3153]. An alternative method for doing this, which has the additional advantage of avoiding discontinuities near the edge of the cut-out diagonal region, has been implemented in the NMRPipe processing software. This procedure works by temporarily shifting the diagonal to the center of the spectrum so that it can be removed by traditional numerical solvent suppression methods [Marion et al., 1989][Callaghan et al., 1984]. Variations on this scheme can also produce a complementary spectrum that contains only the absorptive diagonal signals. A complete description of these processing schemes along with example NMRPipe processing scripts can be found at http://spin.niddk.nih/bax-apps/NMRPipe/diag.
An example of part of the COSY spectrum of human ubiquitin, from which the diagonal has been removed in the above manner, is shown in Figure 1. A second spectrum containing only the absorptive diagonal signals is also derived (not shown). Fitting of a single isolated, in-phase diagonal multiplet is rather insensitive to initial line width or multiplicity, and can be used to obtain the intrinsic signal amplitude. This fit can be repeated for several diagonal signals in order to establish reproducibility. In the ubiquitin case, fitting six different diagonal signals indicates that the amplitude could be determined to an accuracy of better than 5%. Alternatively, the integrated intensity of the entire diagonal subspectrum, or a fraction thereof, divided by the number of spins contributing to this diagonal can be used.
Figure 2 shows how the best-fit active J coupling for the ubiquitin Lys6 Hb¢¢-Ha COSY cross peak depends strongly on this intrinsic intensity. Figure 2A is the experimental multiplet, whereas Figures 2B-D are best-fitted simulated multiplets, where the intrinsic amplitude has been at its true value (Fig. 2B) and at 10 and 100 times larger values (Fig. 2C,D). The goodness of the fit (c2) for the multiplet shown is nearly indistinguishable, yet the magnitude of the active coupling decreases by almost an order of magnitude when the intrinsic intensity, I, is increased from 1 to 100. To a good approximation, in the limit where the line width is larger than the active J coupling, the best fitted coupling scales with the square root of the intensity, whereas the goodness of the fit remains comparable (Fig.2). This confirms that in the absence of amplitude information it is not possible to obtain an accurate J coupling from fitting a single antiphase COSY cross peak.
So, passive couplings can be kept either as fixed or as adjustable parameters during the fitting procedure. If kept variable, the accuracy of the resulting best-fitted passive couplings is poor, however, as the effect of an unresolved passive coupling is similar to that of the fitted natural line width parameter. Convergence of the least-squares minimizer is fastest when estimated approximate values for the passive couplings are entered as fixed non-variable parameters. This is typically the method in which we use the fitting procedure when no (partially) resolved passive splittings are observed in the cross peak.
The graphical interface for coupling extraction is shown in Figure 3. The window in Figure 3A shows a small region of the COSY spectrum of ubiquitin. The boxed region shows the group of multiplets manually selected for analysis. The window in Figure 3B contains the parameters of a fitted model for the first of five interactively selected signals. Parameters that are variable during the fit are marked by black checkboxes. The selected spectral region, corresponding model, and residual spectrum are shown together in a third window (Fig. 3C, 3D, and 3E). If multiple identical passive couplings are present, as for example in the case of the three Hb-Hg couplings in a Hb-Ha cross peaks of threonine residues, the multiplicity for a single passive coupling can be set to ‘3’ in the parameter window, rather than defining three independent couplings. Similarly, when fitting a cross peak involving a methyl group, the intensity can be multiplied by three. The user interface currently allows for up to five multiplets with up to three independent passive couplings per dimension to be fit simultaneously. For larger numbers of cross peaks, the accuracy and convergence of fitted parameters decrease. As shown in Figure 3, not all cross peaks present in the selected window region (Fig. 3C) need to be included in the fit. To illustrate this feature, only five of the eight cross peaks present in Figure 3C were included, and the additional cross peaks remain present in the difference spectrum (Fig. 3E).
When fitting spectra recorded in a dilute liquid crystalline phase [Tjandra, 1997 #195], frequently the number of passive couplings will be much larger than three. However, from a practical perspective, only the largest passive couplings play a role in the fitting procedure and three passive couplings are more than sufficient for the wide variety of spin systems we have studied so far.
Figure 4 compares values for all 58 3J(Ha,Hb) couplings previously measured with the HA(CA)HB experiment [Grzesiek, 1995 #90] with those obtained from the new fitting procedure. On average, J values derived from the HA(CA)HB spectrum underestimate the true coupling if no correction is made for the finite life time of the spin state of the coupling partner [Grzesiek, 1995 #90]. Because the new fitting procedure is not affected by spin-flips of the passive spin, which merely affect the fitted line-width, fitted J values tend to be larger. Overall, agreement is good (Pearson’s correlation coefficient R=0.93) and comparable in quality to that between HA(CA)HB data and heteronuclear E.COSY measurements for ubiquitin (data not shown). This indicates that the present approach for measuring these couplings is equally robust and therefore quite accurate. Any given coupling can be measured twice, from each of the two corresponding cross peaks. Reproducibility is invariably found to be quite high, with a root-mean-square difference of 0.7 Hz over the entire set of Ha/Hb cross peaks (N=78), indicating a random uncertainty of ±0.5 Hz in individual fits.
One detail that may require particular attention is the assumption of uniform intrinsic intensity. In order for this assumption to be valid, the spin system must be fully relaxed at the start of the COSY pulse sequence. Alternatively, two spectra with different interscan delays may be used, such that incomplete T1 relaxation rates can be accounted for. Also, water suppression in phase-sensitive COSY spectra can be a problem as solvent presaturation may unevenly affect the longitudinal 1H magnetization through spin diffusion involving spatially proximate exchangeable protons, or protons resonating in the immediate vicinity of the H2O signal. For this reason, we prefer to record the COSY spectrum in D2O. In principle, double quantum filtering may also be used to attenuate the H2O signal [Piantini, 1982 #3151]. However, this decreases the inherent sensitivity of the COSY experiment two-fold and does not solve the dynamic range problem because the water signal remains present in individual transients. Also, it removes the amplitude information contained in the diagonal and therefore makes our fitting procedure less straightforward. In nucleic acids, partial exchange of base protons with solvent deuterons can result in erroneous amplitudes and thereby introduce errors in the derived couplings. The same, of course, applies to cross peaks to amides for proteins dissolved in D2O.
The fitting procedure is based on the use of eq [1], i.e. on the assumption of first order, weakly coupled spectra. Fitting of cross peaks very close to the diagonal is affected by the diagonal removal routine described above, and therefore does not yield reliable results. In contrast, simulated results indicate that fitting of A-X or B-X cross peaks in an ABX spin system yields reliable results provided that |dA - dB| > ~ 2 JAB. So, although this approach is less rigorous than that developed by Schmidt [Schmidt, 1998 #3206] which takes the strong coupling into account in the model function, ACME is fully adequate for most cases of practical interest.
The ACME program also contains the option to include the effect of delays preceding the t1 and/or t2 evolution period. Such delays may be desirable to allow dephasing of the rapidly decaying signals of bicelle or phage liquid crystal contributions.
The method described here is particularly useful for measurement of 1H-1H couplings in molecules that are weakly aligned in a dilute liquid crystalline phase. These frequently give rise to completely unresolvable cross peak multiplets with more than half a dozen passive couplings, that are difficult to analyze accurately by the E.COSY method. The principal disadvantage of the ACME method is the absence of sign information for the coupling involved. Several novel heteronuclear E.COSY-like methods have been presented recently that permit experimental measurement of both the sign and magnitude of 1H-1H couplings [Otting, 2000 #2367; Peti, 2000 #2332]. However, without isotopic enrichment, such experiments are generally not applicable to macromolecules. If a reasonably accurate initial structure is available, this frequently can be used to determine the sign of the coupling. Alternatively, the structure calculation can use the absolute values of 1H-1H dipolar couplings as input restraints [Tjandra, 2000 #2068].
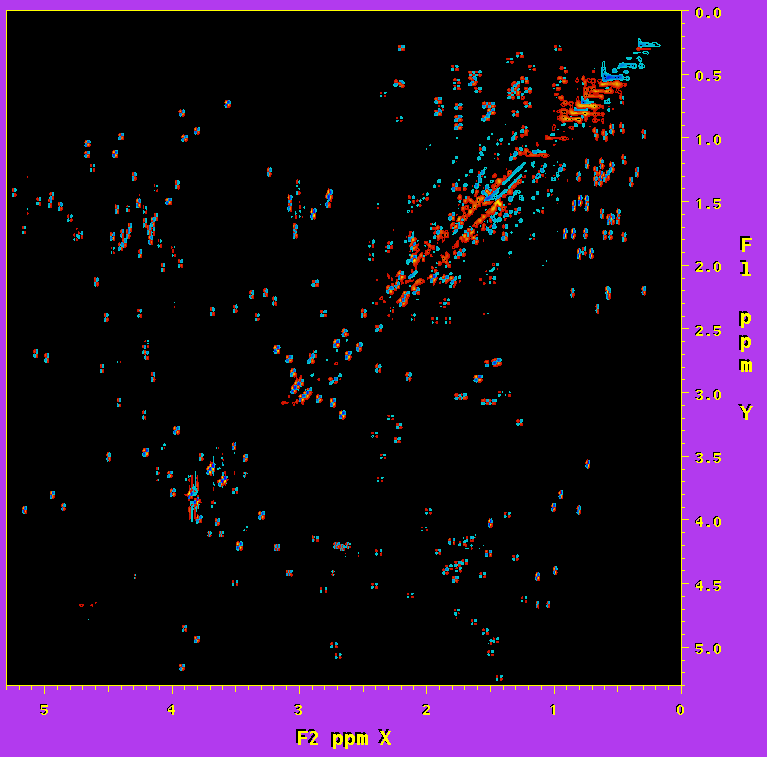
Figure 1. Region of the 800 MHz phase-sensitive COSY spectrum of human ubiquitin, recorded in D2O, from which the dispersive diagonal signals have been removed using the procedure described in the text.
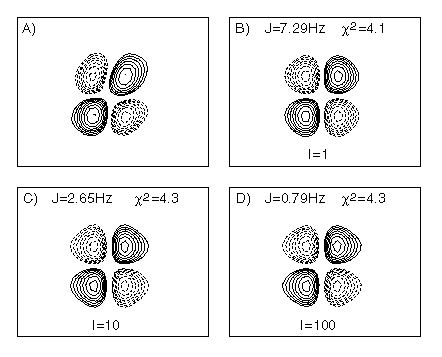
Figure 2. The Hb-Ha cross peak of ubiquitin Glu16 in the 800 MHz COSY spectrum. (A) experimental data, (B) best-fit simulated data using the correct intensity factor (I=1), and best fits when the intensity was constrained to be ten-fold (C) or 100-fold (D) larger. Note that the goodness of the fit (c2) is essentially the same for the three simulated spectra.
|
|
||
Figure 3. Graphical interface for coupling extraction. (A) Region of the COSY spectrum with a zoom-box marking the spectral region on which multiplet fitting is to take place. (B) Parameter window that defines adjustable (dark check box) and fixed parameters to be used in the fit for the first of up to five cross peaks. For passive couplings, the ‘A^1’ mark indicates the number of passive couplings of this size to be included in the simulation; i.e., for passive coupling to a methyl group this value is changed to ‘A^3’. Similarly, the intensity parameter ‘Hi*1’ can be adjusted to ‘Hi*3’ for cross peaks involving a methyl group. Experimental data (C), best fitted data (D) and difference spectrum (E) when only the signals of the five rightmost cross peaks are entered in the parameter window and included in the fit optimization.
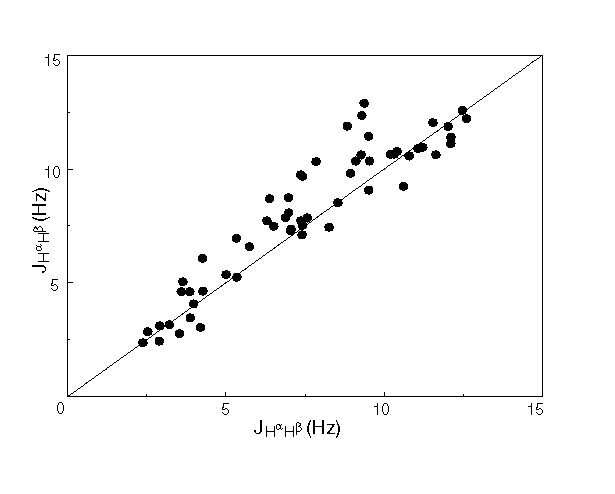
Figure 4. Comparison of 58 3J(Ha,Hb) couplings in human ubiquitin, measured with the new fitting procedure (vertical axis) versus those measured previously with the HA(CA)HB experiment. The systematically smaller value for the HA(CA)HB derived couplings is attributed to the effect of passive spin-flips, which do not affect the result of the multiplet fitting procedure.